“Isso vai mudar a medicina … Vai mudar tudo”, afirma cientista sobre descoberta da IA do Google DeepMind
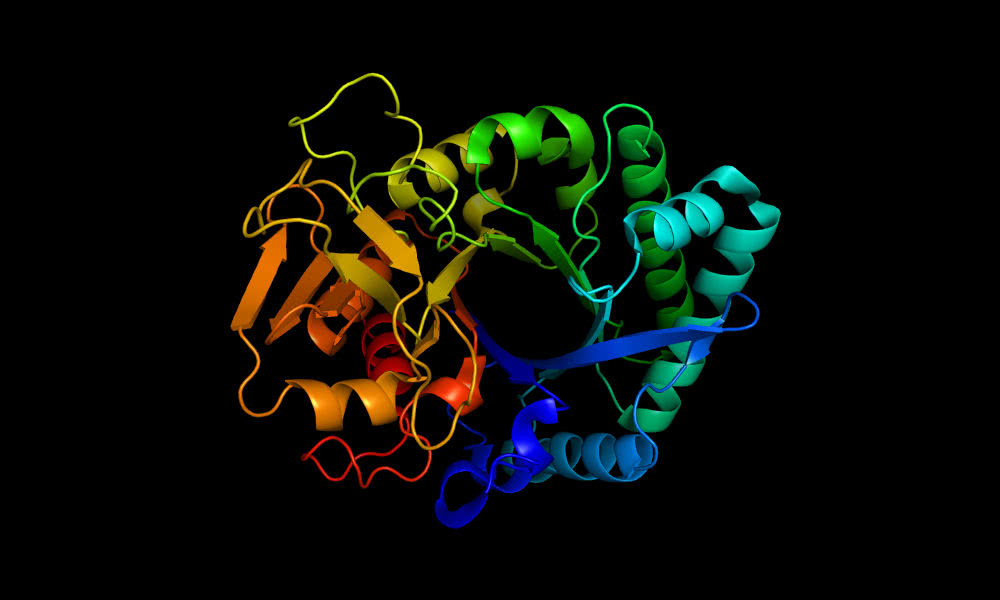
Uma rede de inteligência artificial (IA) desenvolvida pela DeepMind do Google deu um salto gigantesco na resolução de um dos maiores desafios da biologia – determinar a forma 3D de uma proteína a partir de sua sequência de aminoácidos.
O programa da DeepMind, chamado AlphaFold, superou cerca de 100 outras equipes em um desafio bienal de previsão de estrutura de proteína chamado CASP (na sigla em inglês), que significa Avaliação Crítica de Previsão de Estrutura. Os resultados foram divulgados no dia 30 de novembro, no início da conferência — realizada via internet este ano — que avalia os resultados do exercício, de acordo com a Nature.
“Isso foi muito importante”, disse John Moult, um biólogo computacional da Universidade de Maryland em College Park, co-fundador aa CASP em 1994 para melhorar os métodos computacionais para prever com precisão as estruturas de proteínas. “De certo sentido, o problema está resolvido.”
A capacidade de prever com precisão as estruturas das proteínas a partir de sua sequência de aminoácidos seria um grande benefício para as ciências biológicas e a medicina. Isso aceleraria enormemente os esforços para entender os “tijolinhos” que formam as células e permitiria a descoberta de medicamentos mais rápida e avançada.
AlphaFold ficou no topo da tabela no último CASP em 2018, o primeiro ano em que a DeepMind, com sede em Londres, participou. Mas, neste ano, a rede de aprendizagem profunda (deep learning) do equipamento estava muito além das outras equipes e, dizem os cientistas, teve um desempenho tão surpreendente que poderia anunciar uma revolução na biologia.
“É uma virada de jogo”, diz Andrei Lupas, biólogo evolucionista do Instituto Max Planck de Biologia do Desenvolvimento em Tübingen, Alemanha, que avaliou o desempenho de diferentes equipes no CASP. AlphaFold já o ajudou a encontrar a estrutura de uma proteína que incomodou seu laboratório por uma década, e ele espera que isso altere seu funcionamento e as questões que enfrenta. “Isso vai mudar a medicina. Isso mudará a pesquisa. Isso mudará a bioengenharia. Vai mudar tudo”, acrescenta Lupas.
Em alguns casos, as previsões de estrutura do AlphaFold eram indistinguíveis daquelas determinadas usando métodos experimentais “padrão ouro”, como cristalografia de raios-X e, nos últimos anos, microscopia crioeletrônica. AlphaFold pode não evitar a necessidade desses métodos laboriosos e caros — por enquanto — dizem os cientistas, mas a IA tornará possível estudar os seres vivos de novas maneiras.
O problema de estrutura
As proteínas são os “tijolinhos” dos quais a vida é feita, responsáveis pela maior parte do que acontece dentro das células. Como uma proteína funciona e o que ela faz é determinado por sua forma 3D; “estrutura é função” é um axioma da biologia molecular. As proteínas tendem a adotar sua forma sem ajuda, guiadas apenas pelas leis da física.
Por décadas, experimentos de laboratório foram a principal forma de obter boas estruturas de proteínas. As primeiras estruturas completas de proteínas foram determinadas, a partir da década de 1950, por meio de uma técnica em que feixes de raios X são disparados contra proteínas cristalizadas e a luz difratada traduzida em coordenadas atômicas de uma proteína. A cristalografia de raios X produziu a maior parte das estruturas de proteínas. Mas, na última década, a microscopia crioeletrônica se tornou a ferramenta preferida de muitos laboratórios de biologia estrutural.
Os cientistas há muito se perguntam como as partes constituintes de uma proteína — cadeias de aminoácidos — mapeiam as muitas voltas e dobras de sua forma final. As primeiras tentativas de usar computadores para prever estruturas de proteínas nas décadas de 1980 e 1990 tiveram um desempenho ruim, dizem os pesquisadores.
Moult iniciou o CASP para trazer mais rigor a esses esforços. O evento desafia as equipes a prever as estruturas de proteínas que foram resolvidas usando métodos experimentais, mas para as quais as estruturas não foram divulgadas. Moult credita o experimento — ele não o chama de competição — por ter melhorado muito o campo.
O desempenho da DeepMind em 2018 no CASP13 surpreendeu muitos cientistas da área, que há muito tempo é a fortaleza de pequenos grupos acadêmicos. Mas sua abordagem era bastante semelhante à de outras equipes que estavam aplicando IA, diz Jinbo Xu, biólogo computacional da Universidade de Chicago, Illinois.
A primeira iteração de AlphaFold aplicou o método IA conhecido como aprendizado profundo a dados estruturais e genéticos para prever a distância entre pares de aminoácidos em uma proteína. Em uma segunda etapa que não utiliza IA, AlphaFold usa essas informações para chegar a um “consenso” de modelo de como a proteína deve aparentar, diz John Jumper da DeepMind, que está liderando o projeto.
A equipe tentou desenvolver essa abordagem, mas acabou em lugar nenhum. Portanto, mudou de rumo, diz Jumper, e desenvolveu uma rede de IA que incorporou informações adicionais sobre as restrições físicas e geométricas que determinam como uma proteína se dobra. Eles também definiram uma tarefa mais difícil: em vez de prever relações entre aminoácidos, a rede prevê a estrutura final de uma sequência de proteína alvo. “É um sistema um pouco mais complexo”, diz Jumper.
Precisão surpreendente
CASP decorre durante vários meses. Proteínas alvo ou porções de proteínas chamadas domínios — cerca de 100 no total — são liberadas regularmente e as equipes têm várias semanas para enviar suas previsões de estrutura. Uma equipe de cientistas independentes avalia então as previsões usando métricas que avaliam o quão semelhante uma proteína prevista é com a estrutura determinada experimentalmente. Os avaliadores não sabem quem está fazendo uma previsão.
As previsões do AlphaFold chegaram com o nome de ‘grupo 427’, mas a precisão surpreendente de muitas de suas entradas as fez se destacar, diz Lupas. “Eu tinha adivinhado que era AlphaFold. A maioria das pessoas tinha”, diz ele.
Algumas previsões foram melhores do que outras, mas quase dois terços foram comparáveis em qualidade às estruturas experimentais. Em alguns casos, diz Moult, não estava claro se a discrepância entre as previsões do AlphaFold e o resultado experimental era um erro de previsão ou um artefato (um “erro”) do experimento.
As previsões do AlphaFold não batiam com as estruturas experimentais determinadas por uma técnica chamada imagem de ressonância magnética nuclear, mas isso pode ser devido à forma como os dados brutos são convertidos em um modelo, diz Moult. A rede também luta para modelar estruturas individuais em complexos de proteínas, ou grupos, por meio dos quais as interações com outras proteínas distorcem suas formas.
No geral, as equipes previram estruturas com mais precisão este ano, em comparação com o último CASP, mas muito do progresso pode ser atribuído ao AlphaFold, diz Moult. Em alvos de proteína considerados moderadamente difíceis, os melhores desempenhos de outras equipes normalmente pontuaram 75 em uma escala de 100 pontos de precisão de previsão, enquanto AlphaFold marcou cerca de 90 nos mesmos alvos, diz Moult.
Cerca de metade das equipes mencionou “aprendizado profundo” no resumo, resumindo sua abordagem, diz Moult, sugerindo que a IA está causando um amplo impacto no campo. A maioria deles era de equipes acadêmicas, mas a Microsoft e a empresa de tecnologia chinesa Tencent também entraram no CASP14.
Mohammed AlQuraishi, biólogo computacional da Universidade de Columbia em Nova York e participante do CASP, está ansioso para se aprofundar nos detalhes do desempenho do AlphaFold no concurso e aprender mais sobre como o sistema funciona quando a equipe DeepMind apresentar sua abordagem em 1º de dezembro . É possível — mas improvável, diz ele — que uma gama de alvos de proteína mais fácil do que o normal contribuiu para o desempenho. O forte palpite de AlQuraishi é que AlphaFold será transformacional.
“Acho que é justo dizer que isso será muito prejudicial para o campo de predição de estrutura de proteína. Suspeito que muitos deixarão o campo porque o problema central foi sem dúvida resolvido”, diz ele. “É um avanço de primeira ordem, certamente um dos resultados científicos mais significativos da minha vida.”
Estruturas mais rápidas
Uma previsão do AlphaFold ajudou a determinar a estrutura de uma proteína bacteriana que o laboratório de Lupas vem tentando descobrir há anos. A equipe de Lupas já havia coletado dados brutos de difração de raios-X, mas transformar esses padrões aleatórios em uma estrutura requer algumas informações sobre a forma da proteína. Truques para obter essas informações, assim como outras ferramentas de previsão, falharam. “O modelo do grupo 427 nos deu nossa estrutura em meia hora, depois de uma década experimentando de tudo”, diz Lupas.
Demis Hassabis, cofundador e executivo-chefe da DeepMind, diz que a empresa planeja tornar o AlphaFold útil para que outros cientistas possam utilizá-lo. (Publicou anteriormente detalhes suficientes sobre a primeira versão do AlphaFold para outros cientistas replicarem a abordagem.) AlphaFold pode levar dias para chegar a uma estrutura prevista, que inclui estimativas sobre a confiabilidade de diferentes regiões da proteína. “Estamos apenas começando a entender o que os biólogos desejam”, acrescenta Hassabis, que vê a descoberta de medicamentos e o design de proteínas como aplicações potenciais.
No início de 2020, a empresa divulgou previsões das estruturas de um punhado de proteínas SARS-CoV-2 que ainda não haviam sido determinadas experimentalmente. As previsões do DeepMind para uma proteína chamada Orf3a acabaram sendo muito semelhantes a uma determinada posteriormente por meio de microscopia crioeletrônica, diz Stephen Brohawn, neurobiólogo molecular da Universidade da Califórnia, Berkeley, cuja equipe lançou a estrutura em junho. “O que eles conseguiram fazer é muito impressionante”, acrescenta.
Impacto no mundo real
É improvável que AlphaFold feche laboratórios, como o de Brohawn, que usam métodos experimentais para resolver estruturas de proteínas. Mas isso pode significar que dados experimentais de qualidade inferior e mais fáceis de coletar seriam tudo o que você precisava para obter uma boa estrutura. Algumas aplicações, como a análise evolutiva de proteínas, estão definidas para florescer porque o tsunami de dados genômicos disponíveis pode agora ser traduzido de forma confiável em estruturas. “Isso vai capacitar uma nova geração de biólogos moleculares a fazer perguntas mais avançadas”, diz Lupas. “Vai exigir mais reflexão e menos pipetagem.”
“Este é um problema que eu estava começando a pensar que não seria resolvido em minha vida”, diz Janet Thornton, bióloga estrutural do European Molecular Biology Laboratory-European Bioinformtics Institute em Hinxton, Reino Unido, e ex-assessor do CASP. Ela espera que a abordagem possa ajudar a desvendar a função de milhares de proteínas não resolvidas no genoma humano e dar sentido às variações de genes causadores de doenças que diferem entre as pessoas.
O desempenho de AlphaFold também marca um ponto de inflexão para DeepMind. A empresa é mais conhecida por usar IA para dominar jogos como Go e xadrez, mas seu objetivo de longo prazo é desenvolver programas capazes de alcançar uma inteligência humana ampla. Enfrentar grandes desafios científicos, como a previsão da estrutura da proteína, é uma das aplicações mais importantes que sua IA pode fazer, diz Hassabis. “Acho que é a coisa mais significativa que fizemos, em termos de impacto no mundo real.” [Nature]
